A proposed mechanism for vaccine-induced thrombotic thrombocytopenia
In brief...
This study provides an explanation for antibody-induced platelet activation that may contribute to clot formation in patients who received adenoviral vector vaccines against COVID-19.
What did the researchers do?
Samples from patients exposed to either heparin or the AstraZeneca vaccine were collected at the McMaster Platelet Immunology Laboratory for Diagnosis.
- VITT samples were taken from 5 patients (2 females, 3 males) who received a single dose of the AstraZeneca vaccine and developed thrombocytopenia and thrombosis. The mean age of patients was 44 years and mean time from first vaccine dose to sample collection was 28 days. Antibodies against PF4 were detected in all samples.
- HIT samples were taken from 10 patients (5 females, 5 males) who had thrombocytopenia after receiving heparin. Nine out of 10 patients experienced thrombosis. The mean age of patients was 69 years and mean time from heparin initiation to sample collection was 14.3 days. Antibodies against P4F-heparin were detected in all samples.
- Patterns of platelet reactivity in VITT and HITT samples were studied in vitro.
- 70 unique recombinant PF4 mutants—each differing by a single amino acid—were produced to identify the specific amino acid targets critical to binding of VITT antibodies on PF4.
What did the researchers find?
Both VITT and HIT samples had unique patterns of platelet reactivity in vitro:
- In VITT samples, platelet activation was not dependent on heparin but was strongly dependent on PF4 dose. Results indicate that VITT antibodies require PF4 for platelet activation and this is mediated by engagement of FcgRIIa receptors.
- In HIT samples, platelet activation occurred according to two distinct profiles: heparin-dependent (n=4) and heparin-independent (n=6). All HIT samples also activated platelets with the addition of PF4.
Researchers identified 8 surface amino acids considered essential to binding of anti-PF4-antibodies from patients with VITT; 4 of these 8 amino acids corresponded to amino acids on PF4 where heparin usually binds in HIT. This may account for the observation that adding heparin to VITT samples inhibits platelet activation, presumably by displacing VITT anti-PF4 antibodies.
Image
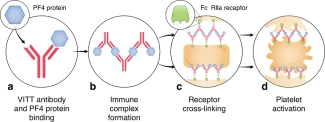
Figure 1: Proposed mechanism for VITT antibodies forming platelet-activating immune complexes (Source Huynh, A., Kelton, J.G., Arnold, D.M. et al. Antibody epitopes in vaccine-induced immune thrombotic thrombocytopaenia. Nature 596, 565–569 (2021).
How can you use this research?
This paper shows for the first time a potential mechanism for platelet activation in a new, rare clotting disease. The findings are a major step forward in the diagnosis and treatment of patients with VITT.
About the research team
This research was led by Dr. Angela Huynh, Department of Medicine, Michael G. DeGroote School of Medicine, McMaster University, Hamilton, Ontario, Canada
This research unit is derived from the following publication(s)
This Research Unit is derived from the following publication:
Huynh, A., Kelton, J.G., Arnold, D.M. et al. Antibody epitopes in vaccine-induced immune thrombotic thrombocytopaenia. Nature 596, 565–569 (2021). https://doi.org/10.1038/s41586-021-03744-4
Author: This Research Unit was written by Dr. Khalid Batarfi, Transfusion Medicine fellow at McMaster University.
Acknowledgements: MCTR receives funding support from Canadian Blood Services (Transfusion Medicine Research Program Support Award), funded by the federal government (Health Canada) and the provincial and territorial ministries of health. The views herein do not necessarily reflect the views of Canadian Blood Services or the federal, provincial or territorial governments of Canada
Keywords: vaccine-Induced immune thrombotic thrombocytopenia, VITT, heparin-induced thrombocytopenia, HIT, epitopes
Want to know more? Contact Ishac Nazy at email: nazyi@mcmaster.ca
A proposed mechanism for vaccine-induced thrombotic thrombocytopenia (PDF)
Published on